

I am currently using Seurat for my scRNA-seq and snRNA-seq analysis (10X), and everything seems to be working fine. However, when I tried to create a Scanpy object from a Cellbender-corrected h5 result file, similar to how I create objects in Seurat, I stumbled upon an issue where there are duplicated gene names. I'm unsure about the root cause of this situation.
with h5py.File(file_path, 'r') as f:
gene_names = [x.decode('utf-8') for x in f['matrix']['features']['name'][:]]
gene_ids = [x.decode('utf-8') for x in f['matrix']['features']['id'][:]]
expression_matrix = f['matrix']['data'][:]
indices = f['matrix']['indices'][:]
indptr = f['matrix']['indptr'][:]
shape = f['matrix']['shape'][:]
from scipy.sparse import csc_matrix
expression_matrix = csc_matrix((expression_matrix, indices, indptr), shape=shape)
# Check duplicated gene names in detail
duplicated_genes = pd.Series(gene_names)[pd.Series(gene_names).duplicated(keep=False)]
print(f"Total number of genes: {len(gene_names)}")
print(f"Number of duplicated gene names: {len(duplicated_genes.unique())}")
print("\nDuplicated gene names and their counts:")
print(duplicated_genes.value_counts().head())
Total number of genes: 55487
Number of duplicated gene names: 114
Duplicated gene names and their counts:
Ccl27a 3
Ccl21b 3
Il11ra2 3
Gm35558 3
Pakap 3
Name: count, dtype: int64After extracting the duplicated genes, everything became clear. The issue was caused by a single gene name corresponding to multiple ENSGIDs.
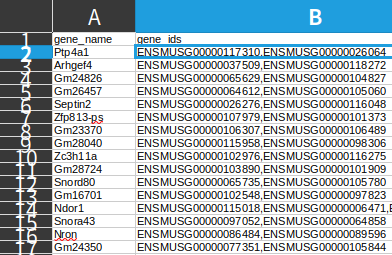
Subsequently, I noticed that during the creation of objects in Seurat, these duplicated genes were appended with suffixes like ".1".
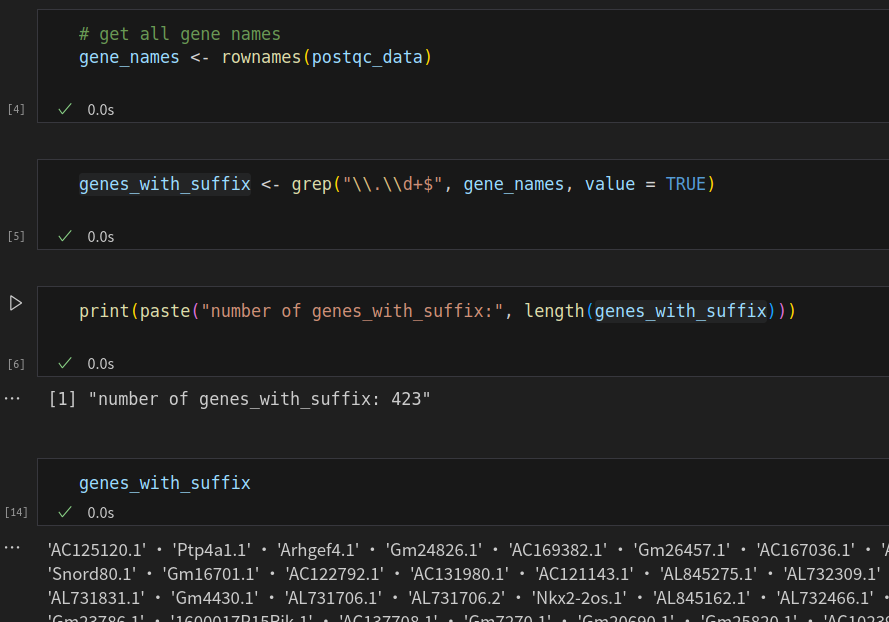
Considering that I will need to perform integration and enrichment analysis using ENSGIDs later, I believe it is better to retain ENSGIDs throughout the analysis. Ideally, both gene names and ENSGIDs should be preserved. It seems that in Python/Scanpy, this can be achieved during object creation using the code below. I am now looking for a solution to achieve the same in R/Seurat.
adata = anndata.AnnData(X=expression_matrix.T)
adata.var_names = gene_names
adata.var['gene_ids'] = gene_ids
Comments NOTHING